Adding a task to the workflow¶
In this tutorial, we will see how to add a new task to the workflow. We will use the example of a task that extract the number of scans from a mzML file, using the pyOpenMS library.
Adding a python script to the workflow executables¶
In cylc-src/bioreactor-workflow/bin/, create a new file named get-scans-number and
paste the following content:
bin/get-scans-number¶#!/usr/bin/env python
import os
import sys
from pathlib import Path
from pyopenms import MzMLFile, MSExperiment
MZML = os.getenv("mzml")
def main():
"""
Usage:
./get-scans-number
Get number of scans from mzML file. `$mzml` shell
environment variable must be set to the path of the file.
"""
exp = MSExperiment()
MzMLFile().load(MZML, exp)
sys.stdout.write(str(exp.getNrSpectra()))
if __name__ == "__main__":
if len(sys.argv) > 1:
sys.stderr.write(main.__doc__)
elif not MZML:
sys.stderr.write("$mzml environment variable not set.\n")
sys.exit()
elif not Path(MZML).exists():
sys.stderr.write(f"mzML file not found: {MZML}\n")
sys.exit()
main()
Make the script executable:
$ chmod +x get-scans-number
Creating a new task in the [runtime] section¶
Open cylc-src/bioreactor-workflow/flow.cylc and add the following task definition at the end:
flow.cylc¶[runtime]
# ...
[[get_scans_number]]
# The task will run in the wf-openms conda environment
# Adding None makes the task appear at the root in the TUI/GUI
inherit = None, CONDA_OPENMS
script = """
echo "The script lauched by this task will extract the number of scans from the mzML file."
get-scans-number > ${output_file}
echo "The number of scans has been saved to ${output_file}"
echo "Number of scans: $(cat ${output_file})"
"""
[[[environment]]]
# The python script will use the $mzml environment
# variable to get the path of the file.
mzml = ${MAIN_RESULTS_DIR}/${RAWFILE_STEM}.mzML
output_file = ${MAIN_RESULTS_DIR}/scans_number.txt
This task will run the get-scans-number script and save the output to a file named
scans_number.txt in the main results directory. This directory
(share/cycle/n/dataflow/) is specific to each cyclepoint n.
Adding the task to the graph¶
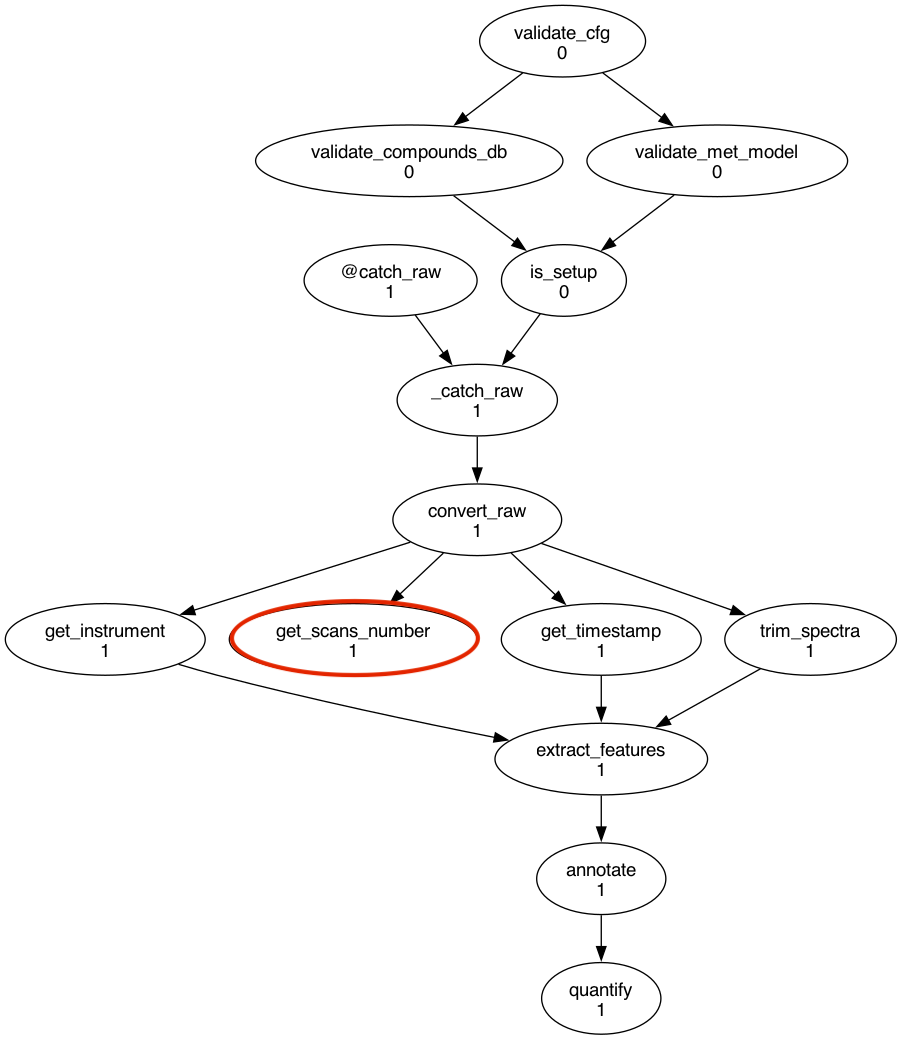
Add a new graph string to the +P1/P1 recurrence, inside the [graph] section of the workflow definition:
flow.cylc¶[[graph]]
R1/^ = validate_cfg => validate_compounds_db & validate_met_model => is_setup
R1/+P1 = convert_raw => get_instrument => extract_features
+P1/P1 = """
is_setup[^] => _catch_raw
@catch_raw => _catch_raw => convert_raw => get_timestamp &
trim_spectra => extract_features => annotate => quantify
convert_raw => get_scans_number
"""
The task will be executed for each cyclepoint (/P1) starting from the second one (+P1). It will run after the convert_raw task as it depends on the mzML file generated by it. No other task depends on the one we just added.
You can check that the task has been added correctly by running:
$ cylc graph bioreactor-workflow 0 1
Testing the new task¶
Install and start a new run of the workflow, and add a mzML file to the raws/ directory. The task should
start immediately after the convert_raw task and generate a scans_number.txt file
in the cylc-run/your_run_name/share/cycle/1/dataflow/ directory.
job.out in logs¶Workflow : bioreactor-workflow/task-added
Job : 1/get_scans_number/01 (try 1)
User@Host: elliotfontaine@MBP-Elliot.local
2024-07-22T14:18:50+02:00 INFO - started
The script lauched by this task will extract the number of scans from the mzML file.
The number of scans has been saved to /Users/elliotfontaine/cylc-run/bioreactor-workflow/task-added/share/cycle/1/dataflow/scans_number.txt
Number of scans: 35
2024-07-22T14:18:52+02:00 INFO - succeeded